One of the healthiest things science can do is make a beautiful dataset less beautiful.
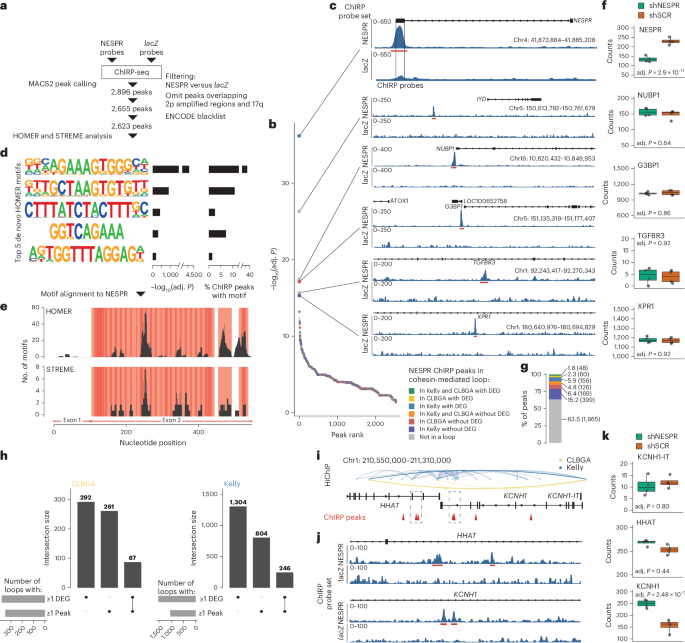
A new paper in *Nature Biotechnology*, published on 15 May, examines common methods used to map where long noncoding RNAs appear to bind across the genome. These RNAs are real and biologically important, but many are present at very low copy number inside cells. That has made one result pattern suspicious: experiments reporting thousands of confident chromatin-binding sites for individual lncRNAs that may exist at only a handful of copies per cell.
The authors studied the chromatin interactome of NESPR lncRNA in cells with varying endogenous expression and performed a meta-analysis across dozens of human and mouse RNA-chromatin interaction datasets. Their central finding is technical and consequential. Many reported peaks may arise when DNA fragments are spuriously recovered because the ends of those fragments partially complement the probes used in pull-down assays such as ChIRP-seq, CHART-seq and RAP-seq. In other words, the experiment can make the probes look as if they found RNA-guided genomic destinations, when at least some signal comes from probe-DNA behaviour.
This does not mean long noncoding RNAs do nothing, or that every chromatin interaction is imaginary. Xist and other well-studied cases still need careful treatment. The narrower claim is more useful: for many trans-acting lncRNAs in mammalian-cell studies, thousands of reported binding regions may be artifacts unless the right controls are present.
The paper also lands as a cultural warning. The accompanying Springer Nature community post says chromatin maps were often a final figure panel, the satisfying bridge between an RNA perturbation and a proposed regulatory mechanism. That is exactly where an artifact can become a story.
The practical takeaway is not cynicism. It is better experimental hygiene: stronger controls, more sceptical interpretation of low-abundance RNAs with vast binding maps, and quality checks before using peaks as mechanistic proof. In biotech terms, this is not a product story. It is a map-cleaning story, and cleaner maps are how future therapeutic biology avoids expensive wrong turns.
There is a broader AI-age lesson in that. As automated literature mining and model-assisted biology make it easier to connect papers into plausible mechanisms, the quality of the underlying assay becomes more important, not less. A model can summarise a map; it cannot rescue a map that was partly produced by the experimental handle used to pull it out of the cell.